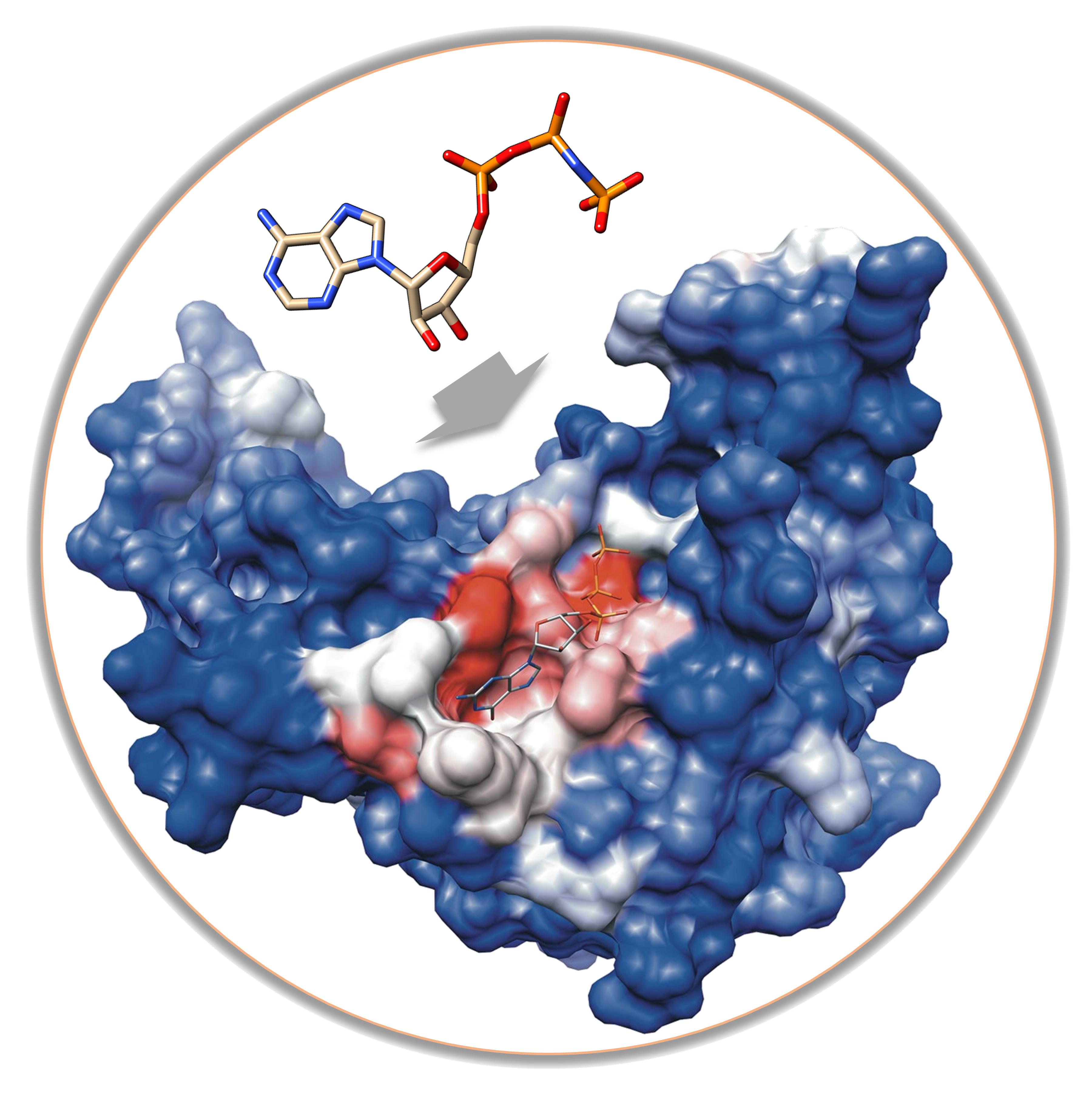
 Docking
Docking
“Predict binding mode of ligand(s) with your protein target.”
Predicting the binding mode of molecules is one of the most common techniques in the computer-aided drug design process.
The known binding mode of an active molecule could be next utilized in
further steps of drug development (e.g. ligand optimization).
Our SilDrug tool can help you in this task by using the AutoDock Vina
docking engine supported by our Open Drug Discovery Toolkit (ODDT)
(Wojcikowski 2015).
More information...
About the tool
AutoDock Vina is open-source, fast, and the most widely used docking software, whereas the ODDT, is our free and open-source tool that handles many aspects of preparation, management, and analysis results of the molecular docking simulations process. Our SilDrug web-based interface will help you relatively easily prepare and run your molecular docking simulation. However, keep in mind that correctness of your input files is very important for the success of running simulations, so please prepare them carefully. To run our SilDrug docking protocol you may need some additional software to properly prepare your input files containing receptor and ligand(s). As our docking engine is AutoDock Vina you may also use any of the software suggested in the tool documentation.
Before running molecular docking simulations first, you need to have a 3D structure coordinates file of your protein target (in PDB format). However, before uploading your files into the docking protocol you should pay attention to the following issues:
- (1) Prepare receptor
- (2) Introduce information about the putative binding site location
- (3) Prepare ligands
(1) Preparing your receptor
To successfully run docking simulations you need first carefully complete the following tasks:
- Check carefully if all atoms of residues creating your putative binding pocket (e.g. active site) for ligand(s) are available in your structure file. If not, they have to be modeled first!
- The missing atoms/residues which are part of your putative binding site for ligand(s) (e.g. active site) should be rebuilt/modeled first
- Check if any of the residues creating your bind pocket has an alternate location. If yes you should decide which one may be appropriate for your ligand binding (e.g. active/inactive conformation) and retain only that one. Alternatively, you may prepare two different simulations for each of the locations (conformation) of the residue
- All other molecules like water molecules, ions, ligands, etc. should be removed from the structure unless they may be important for a ligand binding (e.g. metal ions in active site)
- All hydrogens atoms should be added to the protein
- If your binding pocket is created by residues of one protein chain, you should upload only this chain.
You may prepare your file according to the above requirements using any of the standalone, free protein visualizing software like PyMOL or Chimera UCSF. You may also use web tools like FirstGlance in Jmol to identify regions with problems and missing residues, MolProbity or PDB-Tools Web to clean your PDB file, while protons may be added by PDB Utility Servers or by using an option on our web-tool during uploading your structural file. In the latter case, protons will be added by Open Babel software.
(2) Get knowledge of where your binding site is located.
- (1) You may use prediction results from our Binding Site Assessment tool - Kalasanty (file called “pocket0.mol2”).
- (2) If your protein structure has been resolved in the complex with ligand (e.g. substrate analog, known inhibitor, etc.), you may extract this ligand structure and upload it to our server in the “AUTO LIGAND” option. The protein residues located around the uploaded ligand structure will be automatically selected as your binding pocket for molecular docking.
- (3) Finally you may use the advanced option and manually introduce the center of the docking area (e.g. geometrical center of your binding pocket) coordinates (X, Y, Z) together with box dimensions (A, B, C) in Å, that should surround residues of your ligand binding site.
Accurate information about your binding site location for your ligand(s) is crucial to perform successful molecular docking simulation. On our web server, you have three options to complete this task.
(3) Preparing your ligands
Your ligands should be uploaded in the SDF format. You may find your ligand structure ready for docking in the ZINC database or our version of the European Chemical Biology Database available here. If you would like to prepare ligand structures by yourself please go following this tutorial.