 Pharmacophore
Pharmacophore
“Evaluate your molecules by ligand-based approach”
Comparison of small molecules is a common component of many cheminformatics workflows, including the design of new compounds and libraries as well as side-effect predictions and drug repurposing.
DeCAF is a solution for comparing and assessing compounds based on pharmacophore features (Stepniewska-Dziubinska et al. 2017). DeCAF offers a unique measure of compounds similarity based on their physico-chemical characteristics, abstracting from their chemical structure. Weights in the model can be manually modified to introduce expert knowledge. More information...
About the tool
DeCAF allows large-scale comparisons of low-molecular weight compounds, creating extensive pharmacophore alignments, and thus building models for assessing the activity of chemical molecules without knowledge about the structure of their complexes with the receptor.
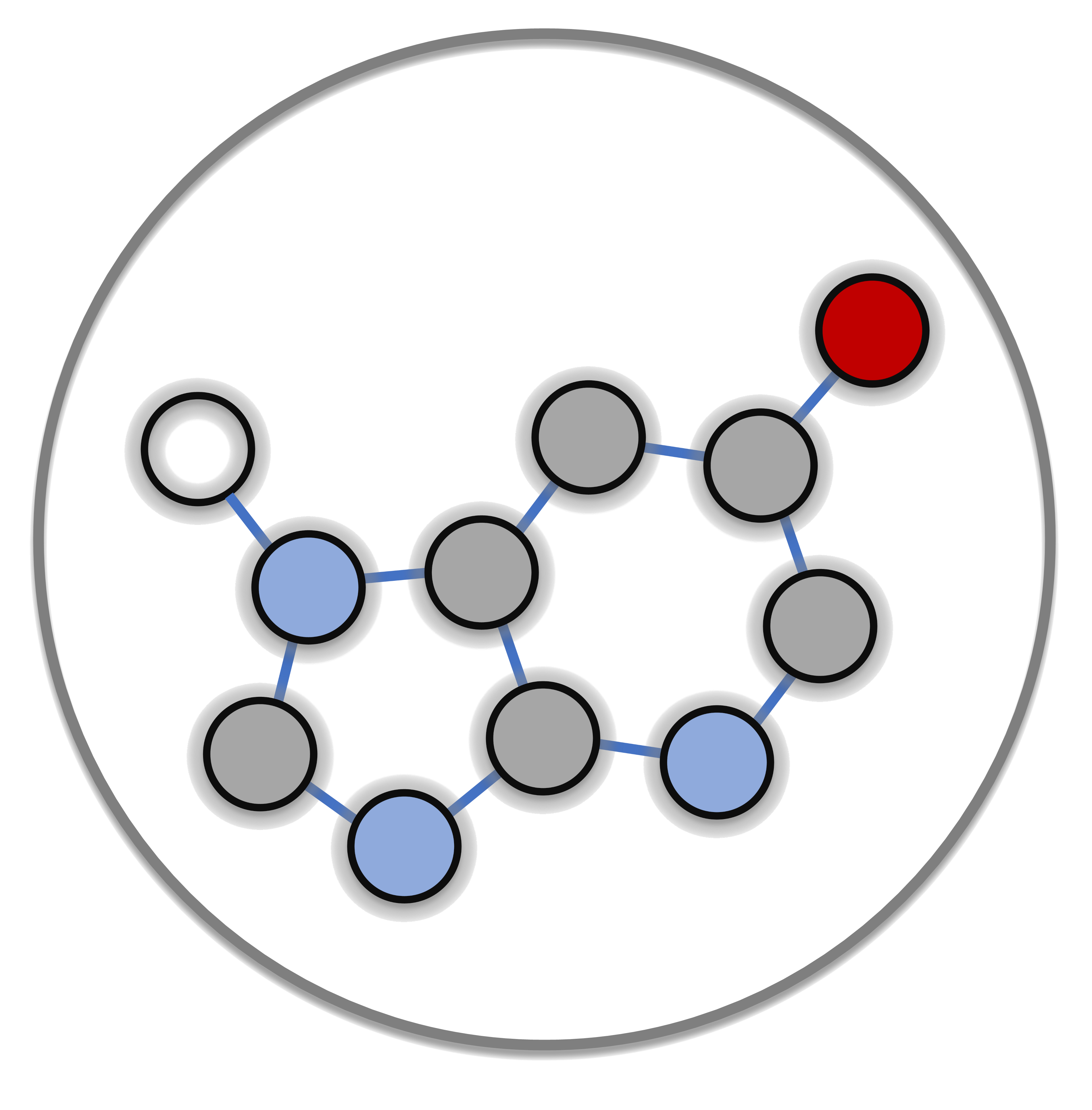
Methods that utilize 3D information depend on multiple conformer generation steps, which are computationally expensive and can greatly influence their results, whereas our open-source software called DeCAF (Discrimination, Comparison, Alignment tool for 2D Pharmacophores), which augments molecule representation with spatial and physicochemical properties while simultaneously avoiding conformer generation.
Our approach describes a molecule as an undirected graph in which the nodes correspond to atoms with pharmacophoric properties and the edges of the graph represent the distances between features. This approach combines the benefits of a conformation-free representation of a molecule with additional spatial information.
If you have a set of active compounds you may use DeCAF using three different modes of action:
- (1) COMPARE - easy and fast compare two sets of ligands to a reference set;
- (2) MODEL - to create your model of pharmacophoric properties based on your active compounds;
- (3) FILTER - filter set of molecules that fulfill the criteria of your pharmacophore model.
Depending on the mode of action which you will choose during job submission you may need different input files/data.
(1) COMPARE
Input
If you just want to compare a new set of molecules to the reference set (active) you need two sets of molecules (evaluated and reference) in smiles format saved in files with the .smi or .ism extension.
Output
- CSV file containing matrix scores (compound similarity) calculated for all molecules from the reference set vs all molecules from the testing set.
- PNG files visualize all comparisons (molecules from the test set vs molecules from the reference set)
(2) MODEL
Input
To create your pharmacophore model use a set of smiles of your active compounds. Your file should have a .smi or
.ism extension.
Cut-off parameter: Describe how restrictive your model should be according to the common features found in
your ligands set. Higher value – more restrictive. The default value is set to 0.7.
Output
- summary.txt - file containing details about your model(s)
- model_[no].json - file(s) containing your model(s)
- visualization - directory containing png files containing graphs representing your model(s)
(3) FILTER
Input
If you would like to filter your set of new compounds according to the previously prepared pharmacophore model you
need (a) file with a set of smiles of your compounds (.smi or .ism extension), and (b) your previously
prepared model by DeCaF (with .json extension).
Cut-off parameter: Describe how restrictive your model should be according to the
common features found in your ligands set. Higher value – more restrictive. The default value is set to 0.7.
Output
- CSV file containing molecules ID that fulfilled the criteria of your pharmacophore model, score value and cost/penalty