 Binding Site Assessment
Binding Site Assessment
“Finding druggable pockets on your protein surface.”
Rational drug design aims to discover new drug-like molecules, but still, most of the structure-based techniques require knowledge about the exact location of ligand(s) binding site(s). However, if you are not able to locate your binding pocket, deep learning methods implemented in the SilDrug can perform this task for you. More information...
About the tool
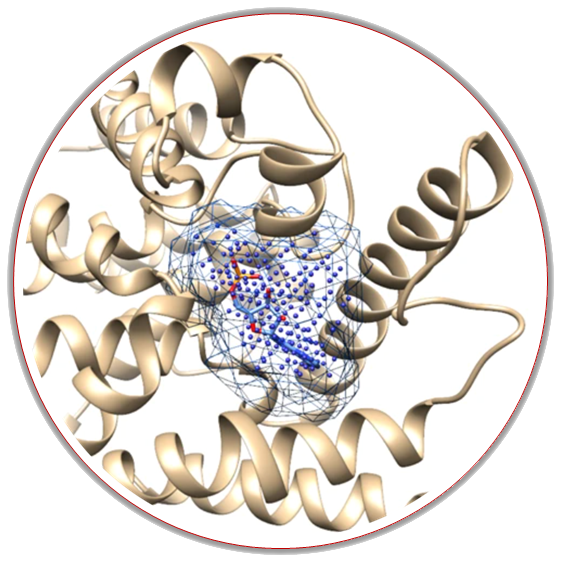
Kalasanty (Stepniewska-Dziubinska et al. 2020) is a deep learning-based approach for finding binding pockets, inspired by semantic image segmentation. Our model, Kalasanty, takes protein structure as input, and automatically converts it to a 3D grid with features, and outputs probability density – each point in the 3D space has an assigned probability of being a part of a pocket. Kalasanty is based on U-Net – a state-of-the-art neural network architecture for semantic segmentation, originally developed for 2D medical images. We adopted the U-Net to process 3D protein structures and provided the model with input relevant to the task of identifying binding cavities.
DeepPocket (Aggarwal et al. 2021) is a novel and comprehensive framework based on a multistep approach to get the final pocket location and 3D shape prediction from the input protein structure. The DeepPocket utilizes 3D convolutional neural networks for the rescoring of pockets identified by a geometry-based software called Fpocket (Le Guilloux et al. 2009) and further segments these identified cavities on the protein surface, which has been shown that outperforms all state-of-the-art methods at identifying and ranking druggable binding sites.
Predictions of your binding site(s) location(s) should be available in minutes (depending on which algorithm you choose, your protein size, and our service occupancy).
Kalasanty will give you output as a .cube file, which can be later analyzed in molecular modeling software, and as a .mol2 file which contains parts of your protein that form pockets to easily visualize it or directly use as a receptor for molecular docking approach.
DeepPocket additionally to the *.mol2 file containing parts of your protein that forms binding sites will give you more details about the score, druggability, size, solvent accessible surface, etc. which you may download and analyze outside the SilDrug together with the scripts which you may use to visualize by PyMol or VMD software.
The predictions by both methods are also ready to visualize directly in the SilDrug service. You will find appropriate buttons on the results webpage which will redirect you directly to implemented Mol* 3D Viewer. The predicted binding pockets will be then presented as sticks, whereas the rest of the protein as cartoons.
Input
Option 1
If you would like to upload the coordinates file of your protein structure (in PDB format) please remember that:
- all other molecules like water molecules, ions, ligands, etc. should be removed from the structure;
- all hydrogen atoms should be added to the protein;
- we also recommend uploading only one chain of your protein structure.
You may prepare your file according to the above requirements using any of the standalone free protein visualizing software like PyMOL or Chimera UCSF. You may also use web tools like MolProbity or PDB-Tools Web to clean your PDB file, while protons may be added by PDB Utility Servers.
Option 2
If you decide to use the PDB ID (e.g. 1PJK) of your protein structure please provide also chain identifier(s) which should be used by the server. The PDB file will be automatically fetched from RCSB and prepared: only atoms with the specified chain(s) ID will be considered, all non-protein molecules will be deleted (water molecules, ligands, ions, etc.), and protons will be added.
Output
Binding site prediction results will be available to download as an archive, compressed file results.tar.gz. After downloading and unpacking you may find inside:
KALASANTY:
- (1) Your input structure in PDB format
- (2) A Gaussian Cube File Format (.cube) file containing volumetric properties of your predicted binding site that can be employed in molecular modeling software
- (3) Atomic coordinates file in the Tripos molecular structure format (.mol2), which contains only parts of the protein that form predicted pocket
DeepPocket:
- (1) Your input structure in PDB format
- (2) A Gaussian Cube File Format (.cube) file containing volumetric properties of your predicted binding site that can be employed in molecular modeling software
- (3) Atomic coordinates file in the Tripos molecular structure format (.mol2), which contains only parts of the protein that form predicted pocket
- (4) *_info.txt text file containing outputs information about different cavities parameters calculated for your protein
- (5) *_PYMOL.sh and *_VMD.sh bash scripts which visualize your results directly in PyMol or VMD software on your computer.
For more information please visit the DeepPocket project GitHub repository
You may also visualize your results directly on our server by using the “VISUALIZE” button on the results page. Your protein will be shown as a cartoon representation, and residues creating predicted binding sites will be shown as sticks in PDB Viewer.